by Matthew B. Zarraga, DO; Nicole Swenson, DO; and Brad Glick, DO, MPH, FAOCD
Dr. Zarraga is the chief dermatology resident at Wellington Regional Medical Center in Wellington, Florida; Dr. Swenson is a research fellow at the Center for Clinical and Cosmetic Research in Aventura, Florida; and Dr. Glick is the program director at Wellington Medical Center and Skin and Cancer Associates in Florida.
Funding: No funding was received for the preparation of this article.
Disclosures: The authors have no conflicts of interest relevant to the content of this article.
Abstract: Granulomatosis with polyangitis, formerly known as Wegener’s granulomatosis, is a multi-system vasculitis that has a variable clinical presentation. Although uncommon, cutaneous symptoms can be the initial presenting symptom of granulomatosis with polyangitis. We present an unusual case of pyoderma gangrenosum followed by a diagnosis of granulomatosis with polyangitis. We also provide a review of current literature on therapeutic options.
Keywords: Pyoderma gangrenosum, granulomatosis with polyangitis, Wegener’s granulomatosis, vasculitis
Granulomatosis with polyangitis (GPA), formerly known as Wegener’s granulomatosis, is one of the antineutrophil cytoplasmic antibody (ANCA) vasculitides that primarily affects the small vessels. Other diseases in this category include microscopic polyangitis and eosinophilic granulomatosis with polyangitis, formerly known as Churg-Strauss.1 First described in 1931 as a variant of polyarteritis nodosa, later advances in laboratory testing allowed for further classification of these diseases based on ANCA subtype. In 2011, Wegener’s granulomatosis was renamed GPA.2 We present an unusual case of GPA that was initially diagnosed as pyoderma gangrenosum.
J Clin Aesthet Dermatol. 2017;10(10):40–42
Case Presentation
A 50-year-old Caucasian man with a past medical history of arthritis presented with a two-month history of progressive, round-oval, deep-seated ulcerations. At initial presentation, the patient developed a nodular abscess in the left groin that occurred after a scratch on his left arm. He presented to the emergency room (ER) and was treated with incision and drainage, which initially relieved the symptoms from the abscess. At that ER visit, a tissue culture was taken, which was later found positive for acid-fast bacilli (AFB) on smear. A few days later, the patient returned to the ER and was admitted to the hospital due to painful, enlarging ulcerations in multiple locations, at which point Dermatology was consulted.
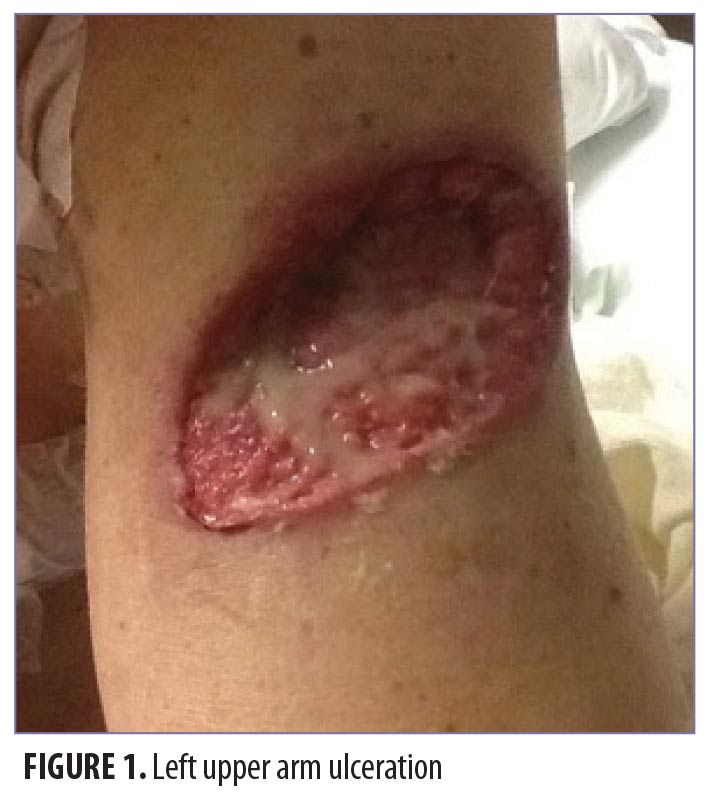
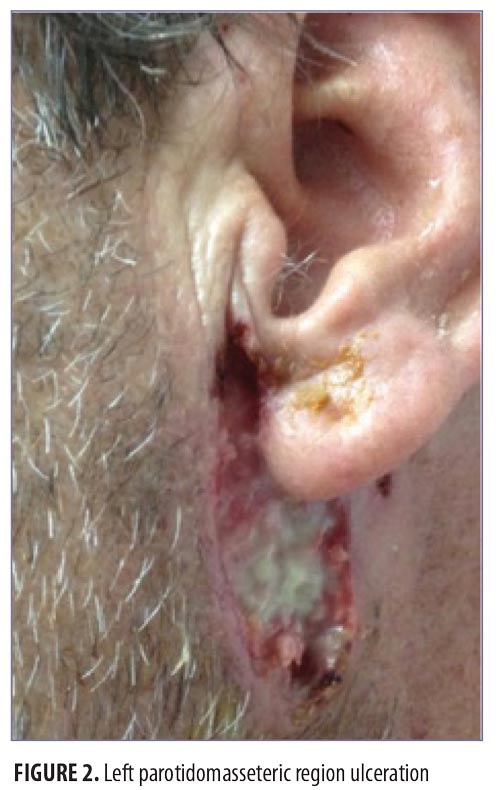
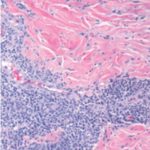
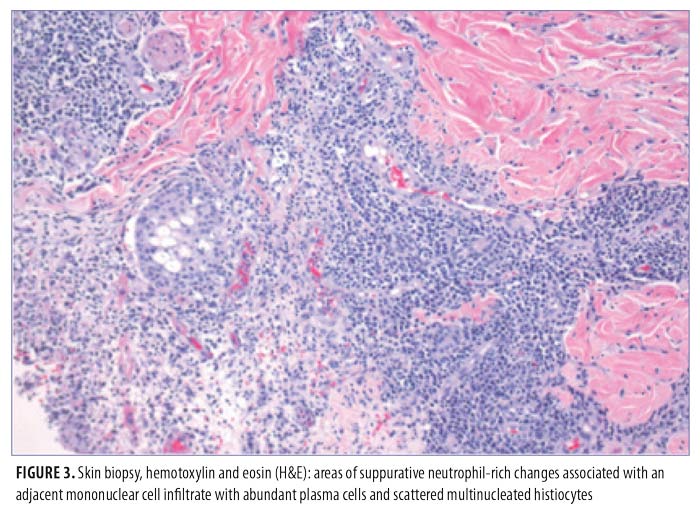
On physical exam, the patient had large, painful ulcerations on the left chest, left upper arm, and left malar and parotidomasseteric regions (Figures 1 and 2). The only associated symptom was intermittent fevers. His medical history was unremarkable except for a reported history of arthritis, and his social history did not reveal any drug abuse. Laboratory tests, including complete blood count, comprehensive metabolic panel, serum protein electrophoresis, urinalysis, urine drug screen, stool, C-reactive protein, rapid plasma reagin, hepatitis panel, and human immunodeficiency virus, were all negative. Likewise, urine, wound, and blood cultures were negative. However, cytoplasmic antineutrophil cytoplasmic antibody (c-ANCA) was positive (1:320). Skin biopsy showed wide areas of suppurative neutrophil-rich changes associated with an adjacent mononuclear cell infiltrate with abundant plasma cells and scattered multinucleated histiocytes (Figure 3). No evidence of a primary vasculitis was noted. Further staining was performed with AFB and Fite techniques (Figures 4 and 5), and these were negative for Mycobacteria and Nocardia respectively. In contrast to the incision and drainage specimen obtained in the ER, the AFB obtained from the biopsy specimen showed no Mycobacteria. Gram, Grocott methenamine silver, and Giemsa were negative for bacteria or fungal elements while the Treponema stain was negative for spirochetes. Kappa and Lambda (Figures 6 and 7) were negative for light chain restriction, and the direct immunofluorescence was negative for immunoglobulin (Ig) G, IgA, IgM, C3, C5b-9, and fibrinogen deposition.
The patient was diagnosed with Wegener’s granulomatosis due to the elevated c-ANCA level, which is 99-percent specific for the disease. Also, other causes of pyoderma gangrenosum, such as leukemia, were ruled out. In the hospital, the patient was treated with intravenous corticosteroids and antibiotics, to which he responded well.
Discussion
ANCA associated vasculitis can affect any caliber blood vessels; however, small vessels are most commonly affected.1 According to the American College of Rheumatology, two of four criteria must be present for diagnosis of GPA, including nasal or oral inflammation, an abnormal chest radiograph, urinary sediment with red blood cells or casts, or granulomatous inflammation on biopsy.3 Interestingly, a positive ANCA titer is not required for diagnosis; however, it will frequently be present. GPA is ANCA-positive for the neutrophil serine protease 3 (PR3); however, PR3-ANCA titers are not used to monitor response to treatment or predict relapse.4 In confirmed cases of GPA, PR3-ANCA is reported to be positive in 84 to 92 percent of cases.5,6
Organ systems most frequently involved with GPA are the upper and lower respiratory tracts and kidneys; however, the skin, heart, peripheral nervous system and eye can also be affected.5 Clinical manifestations of GPA include rapidly progressive necrotizing glomerulonephritis and pulmonary hemorrhage with hemoptysis.1 Cutaneous manifestations have variable morphologies, and might not be the initial presenting symptom. Palpable purpura in leukocytoclastic vasculitis or necrotizing ulcers similar to those seen in pyoderma gangrenosum might be seen.7 The presence of skin lesions has been reported in 9 to 25 percent of cases, and in some reports are as high as 50 percent.5,8,9 As in our case, presentation of pyoderma gangrenosum-like lesions preceeding the diagnosis of GPA have been reported in the literature.10 However, the incidence of GPA presenting as PG-like lesions is infrequent, and has been reported in only one of 166 cases.11 Biopsy alone is not sufficient for diagnosis; however, the histopathology of these lesions show characteristic granulomatous inflammation, neutrophil microabscesses, vasculitis, and necrosis.12 Also, cocaine abuse has been reported to mimic rheumatologic disorders including GPA, and these presentations might be difficult to differentiate from autoimmune-mediated processes.15
In the past decade, several advances in treatment regimens for GPA have improved outcomes. Prior to biologic therapy agents, the standard treatment regimen was remission induction with glucocorticoids and cyclophosphamide, trimethoprim/sulfamethoxazole, and methotrexate. Cyclophosphamide for induction of remission is dosed 2mg/kg daily, can be increased up to 4mg/kg daily, and is given with a slow prednisone taper starting at 1mg/kg daily.5 Rituximab, a monoclonal antibody against CD20, has shown efficacy in remission induction similar to that of cyclosphosphamide. In one study, 64 percent of patients treated with rituximab achieved remission at six months, as compared with 53 percent of patients treated with cyclophosphamide.12 Another 12-month study showed similar sustained remission rates with either cyclophosphamide-azathioprine at 82 percent or rituximab at 76 percent.15
Other treatment modalities are also reported in the literature. Etanercept, a monoclonal antibody against TNF-alpha, did not show efficacy in inducing or sustaining remission when added to the standard treatment regimen in subjects with GPA.16 There are case reports in the literature of complete response to low-dose radiation therapy. A nasal and periorbital mass of GPA was successfully treated with 2 Gy of fractionated radiation therapy.17 However, radiation therapy can only be used in limited disease.
Intravenous immunoglobulin (IVIG) has been reported in successfully treating relapses of GPA, given as daily infusion for four consecutive days each month. One study reported a 59-percent remission rate, when added to stable doses of previous medications for GPA, such as methotrexate. IVIG is not used as a first-line agent, but is important to consider for relapses of GPA.18 Current research is evaluating the efficacy of plasma exchange in treating ANCA-associated vasculitis, such as GPA. Plasma exchange is performed in addition to conventional glucocorticoid and cyclophosphamide therapy.19
Conclusion
Wegener’s granulomatosis, now known as granulomatosis with polyangitis, is a chronic small vessel vasculitis that can present atypically as pyoderma gangrenosum. GPA is an important diagnosis to investigate with ANCA confirmatory testing in subjects with lesions resembling pyoderma gangrenosum, as this might be the presenting symptom of systemic GPA. Sustained remission is the goal for treatment in GPA. The current treatment of choice is glucocorticoid therapy with either rituximab or cyclophosphamide.
References
- Jennette JC, Falk RJ, Bacon PA, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65(1):1–11.
- Falk RJ, Gross WL, Guillevin L, et al. Granulomatosis with polyangiitis (Wegener’s): an alternative name for Wegener’s granulomatosis. Arthritis Rheum. 2011;63(4):863–864.
- Leavitt RY, Fauci AS, Bloch DA, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum. 1990;33(8):1101–1107.
- Finkielman JD, Merkel PA, Schroeder D, et al. Antiproteinase 3 antineutrophil cytoplasmic antibodies and disease activity in Wegener granulomatosis. Ann Intern Med. 2007;147(9):611–619.
- Reinhold-Keller E, Beuge N, Latza U, et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: long-term outcome in 155 patients. Arthritis Rheum. 2000;43(5):1021–1032. Erratum in: Arthritis Rheum. 2000 Oct;43(10):2379.
- Finkielman JD, Lee AS, Hummel AM, et al. ANCA are detectable in nearly all patients with active severe Wegener’s Granulomatosis. Am J Med. 2007;120:643.e9–e14.
- Daoud MS, Gibson LE, DeRemee RA, et al. Cutaneous Wegener’s granulomatosis: clinical, histopathologic, and immunopathologic features of thirty patients. J Am Acad Dermatol. 1994;31(4):605–612.
- Gibson LE, Daoud MS, Muller SA, Perry HO. Malignant pyodermas revisited. Mayo Clin Proc. 1997;72(8):734–736.
- Handfield-Jones SE, Parker SC, Fenton DA, et al. Wegener’s Granulomatosis Presenting as Pyoderma Gangrenosum. Clin Exp Dermatol. 1992;17:197–200.
- Reed WB, Jensen AK, Konwaler BE, Hunter D. The cutaneous manifestations in Wegener’s granulomatosis. Acta Derm Venereol. 1963;43:250–264.
- Zycinska K, Wardyn K, Zielonka TM, Nitsch-Osuch A, Smolarczyk R. Cutaneous changes: an initial manifestation of pulmonary Wegener’s granulomatosis. Adv Exp Med Biol. 2013;755:307–310.
- Jacob SE, Martin LK, Kerdel FA. Cutaneous Wegener’s granulomatosis (malignant pyoderma) in a patient with Crohn’s disease. Int J Dermatol. 2003;42(11):896–898.
- Jiménez-Gallo D, Albarrán-Planelles C, Linares-Barrios M, et al. Pyoderma gangrenosum and Wegener granulomatosis-like syndrome induced by cocaine. Clin Exp Dermatol. 2013;38(8):878–882.
- Stone JH, Merkel PA, Spiera R, Seo P, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363(3):221–232.
- Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363(3):211–220.
- Wegener’s Granulomatosis Etanercept Trial (WGET) Research Group. Etanercept plus standard therapy for Wegener’s granulomatosis. N Engl J Med. 2005;352(4):351–361.
- Wygoda A, Rutkowski T, Sk?adowski K, Hejduk B. Low dose radiotherapy as an effective treatment in a patient with solitary Wegener’s granulomatosis resistant to systemic treatment—case report. Contemp Oncol (Pozn). 2013;17(1):107–111.
- Martinez V, Cohen P, Pagnoux C, Vinzio S, et al. Intravenous immunoglobulins for relapses of systemic vasculitides associated with antineutrophil cytoplasmic autoantibodies: results of a multicenter, prospective, open-label study of twenty-two patients. Arthritis Rheum. 2008;58(1):308–317.
- Walsh M, Merkel PA, Peh CA, et al. Plasma exchange and glucocorticoid dosing in the treatment of anti-neutrophil cytoplasm antibody associated vasculitis (PEXIVAS): protocol for a randomized controlled trial. Trials. 2013;14:73.