J Clin Aesthet Dermatol. 2020;13(1):32–34
 by Francisco Javier Lira-Valero, MD; Nancy Pulido-Díaz, MD; and Marissa de Jesús Quintal-Ramírez, MD
by Francisco Javier Lira-Valero, MD; Nancy Pulido-Díaz, MD; and Marissa de Jesús Quintal-Ramírez, MD
All authors are with the Department of Dermatology at the Instituto Mexicano del Seguro Social and Centro Médico Nacional La Raza in México City, México.
FUNDING: No funding was provided for this study.
DISCLOSURES: The authors have no conflicts of interest relevant to the content of this article.
ABSTRACT: Background. Langerhans cell histiocytosis (LCH) is a neoplasm of the monocyte-macrophage lineage, characterized by clonal proliferation and dissemination of cells that express CD1a+ and CD207. It is a disorder that predominates in childhood. Although the skin is the second most frequently affected organ (30-60%), isolated cutaneous involvement is rare; its frequency does not exceed 4 to 12 percent of cases. Single system-LCH usually has a good prognosis. We describe a case of LCH with isolated cutaneous involvement that presented in an adult patient and was refractory to polychemotherapy.
KEYWORDS: Langerhans cell, histiocytosis, cutaneous
Langerhans cell histiocytosis (LCH) is a neoplasm of the monocyte-macrophage lineage, characterized by clonal proliferation and dissemination of cells that express CD1a+ and CD207+.1,2 Its clinical evolution is highly heterogeneous, ranging from self-healing lesions confined to a one single organ to multisystemic, progressive, and potentially fatal illnes.2 Additionally, LCH is known to cause sequelae and permanent disability.3 We describe a case of LCH with isolated cutaneous involvement that presented in an adult patient adulthood and was refractory to poliquimioterapy.
Case Presentation
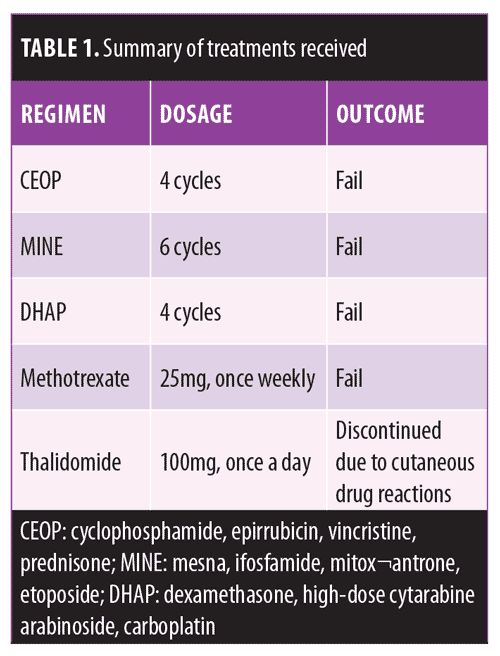
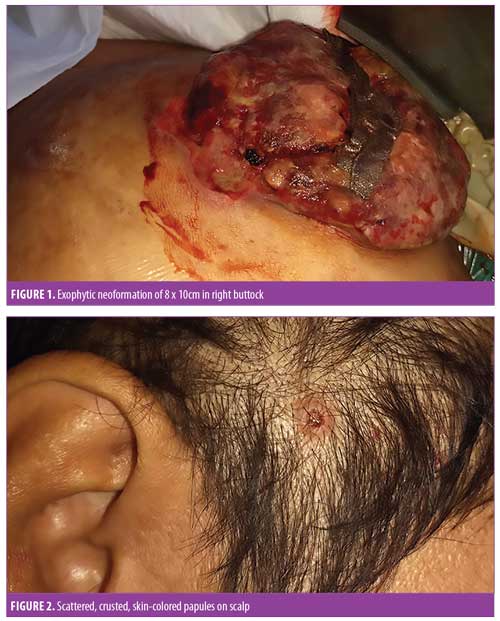
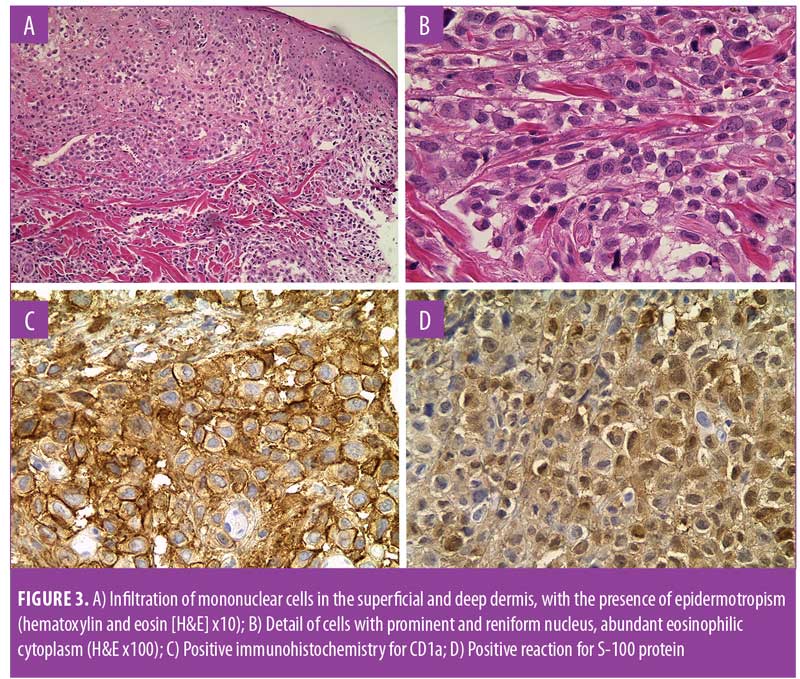
A 53-year-old man with long-standing Type 2 diabetes mellitus (T2DM) was referred to our clinic with a two-year history of exophytic neoformation in the right buttock (Figure 1). Initially, it was classified as anaplastic lymphoma, but based on histopathological and immunohistochemical findings it was reclassified as a cutaneous LCH. The patient underwent various treatment regimens, including systemic chemotherapy and immunomodulatory drugs (Table 1), but remained unresponsive to therapy. A second dermatosis on the scalp was identified at a follow-up visit, which consisted of multiple crusted, skin-colored papules (Figure 2). Histopathological report of both lesions revealed a dense cellular infiltrate in superficial and deep dermis (Figure 3A), constituted by histiocytic cells with reniform nucleus and abundant eosinophilic cytoplasm (Figure 3B). Immunohistochemical staining was positive for CD1a (Figure 3C), protein S-100 (Figure 3D), CD68, and Ki-67 (40%). Laboratory tests and imaging studies did not show evidence of other organs or systems being affected.
The LCH in our patient was classified as refractory to systemic treatment, and the patient underwent hypofractioned palliative radiotherapy (4 Gy in 5 fractions). There was no improvement in pain, bleeding, or discharge after three months of radiotherapy. A functional impairment was reported at the last follow-up visit: the patient had a functional Grade 2 on the Eastern Cooperative Oncology Group (ECOG) score (i.e., patient was ambulatory, up and about more than 50% of waking hours, and capable of all selfcare but unable to work). By this point, the fungating mass in the right buttock had reached a size of 15x15cm. The patient was lost to follow-up.
Discussion
LCH it is a disorder that predominates in childhood; the incidence is 1 to 9 cases per million, and the age range in which the disorder typically presents is 1 to 4 years.1,45 An incidence of 1 to 2 cases per million has been reported in adults.5 Historical designation of “histiocytosis X,” since its conception in 1953, has undergone important changes.1,2,5 In 1987, the Writing Group of the Histiocyte Society reclassified these entities under the name “Langerhans cell histiocytosis.”4 HCL is currently classified within the L-group (Langerhans group), which is distinguished by its clonal mutations that activate the MAPK pathways.4 The clinical stratification has prognostic value and classifies LCH as A) a single system disease (SS-LCH) with unifocal or multifocal involvement or B) a multisystem disease (MS-LCH) with or without involvement of other organs.3,6
The etiology of LCH is uncertain. The hypothesis of immune dysregulation, based on certain alterations of the lesional microenvironment, supports a reactive origin. Specifically, the increased expression of local cytokines such as interleukin (IL)-1, IL-3, IL-4, IL-8, IL-11, granulocyte-macrophage colony-stimulating factor (GM-CSF), interferon-gamma (IFN-gamma), tumor necrosis factor -alpha TNF-alpha), and transforming growth factor-beta(TGF–beta), stimulate the recruitment of other inflammatory cells. In contrast, the presence of certain intrinsic cellular aberrations supports a neoplastic ethology.5 Recurrent mutations in BRAFV600E have been reported in up to 57 percent of LCH specimens, in addition to other alterations in the mitogen-activated protein kinase (MAPK) pathway, such as mitogen-activated protein kinase kinase 1 (MAP2K1), serine/threonine-protein kinase (ARA-F), phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha (PIK3CA), protein interacting with PRKCA 1 (PICK1), and phosphoinositide-3-kinase regulatory subunit 2 (PIK3R2)..2,4
LCH can affect any organ or system, but the most common are bone, skin, and pituitary gland, followed by the liver, spleen, hematopoietic system, lungs, lymph nodes, and central nervous system (CNS).3 Although, the skin is the second most frequently affected organ (30–60%),1,3,4 isolated cutaneous involvement is rare, occurring in only 4 to 12 percent of patients with single system-LCH.7,9 Prognosis is typically good, and relapses occur in less than 10 percent of affected patients.9,10 Cutaneous involvement of LCH can occur anywhere on the body, but the scalp and trunk are the most common sites.1,10 Crusted or scaly papules are the typical cutaneous manifestations of LCH, but the disorder can cutaneously present in any morphology.1,8
In addition to histopathological confirmation, positive immunohistochemical staining is required for CD1a and/or CD207 (Langerin).6 The demonstration of Birbeck granules by electron microscopy is no longer an indispensable criterion.1,3 The lesional tissue is constituted by pathological histiocytes with reniform-like nuclei or in coffee beans; often, other cellular subpopulations are identified, such as B and T lymphocytes, macrophages, eosinophils, and multinucleated giant cells.5
The choice of treatment depends on the site, extent, and severity of the disease. For isolated cutaneous involvement or skin lesions refractory to the treatment of MS-HCL, surgical excision, methotrexate, thalidomide, azathioprine, and phototherapy have all been used. Systemic corticosteroids in combination with chemotherapy drugs are reserved for MS-LCH with or without involvement of other organs, multifocal lesions, or special sites.6
Radiotherapy is a treatment option in selected situations, with the response rate in adult patients ranging from 75 to 100 percent. The main indications for the use of radiotherapy are isolated unresectable lesions, recurrent or progressive lesions, and adjuvant treatment following incomplete resection. Dosages range from 10 to 20Gy.6
Favorable response rates have also been reported with vemurafenib, a selective inhibitor of the B-RAF enzyme. B-RAF (rapidly accelerated fibrosarcoma isoform B) is the most potent activator of the mitogen-activated protein kinase (MAPK) pathway. However, the optimal dose, duration of treatment, and long-term side effects require further investigation.2
Conclusion
LCH in adulthood is uncommon, and its diagnosis requires a high index of suspicion, especially when cutaneous manifestations are the initial presentation. This case report illustrates the unusual skin manifestations that can occur in this disease as well as its unpredictable clinical course. Dermatologists should know how to recognize classic and atypical presentations of this disease, and promptly treat affected patients using a multidisciplinary approach. LCH in adulthood can represent a therapeutic challenge due to the absence of standardized treatment guidelines. The safety and efficacy of pediatric therapies have not been extrapolated to the adult population.
References
- Jezierska M, Stefanowicz J, Romanowicz G, et al. Langerhans cell histiocytosis in children: a disease with many faces—recent advances in pathogenesis, diagnostic examinations and treatment. Postepy Dermatol Alergol. 2018;35(1):6–17.
- Hutter C, Minkov M. Insights into the pathogenesis of Langerhans cell histiocytosis: the development of targeted therapies. Immunotargets Ther. 2016;5:81–91.
- Haupt R, Minkov M, Astigarraga I, et al. Langerhans cell histiocytosis (LCH): guidelines for diagnosis, clinical work-up, and treatment for patients till the age of 18 years. Pediatr Blood Cancer. 2013;60(2):175–84.
- Emile JF, Abla O, Fraitag S, et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood. 2016;127(22):2672–2681.
- Rizzo FM, Cives M, Simone V, Silvestris F. New insights into the molecular pathogenesis cell histiocytosis. Oncologist. 2014;19(2):151–163.
- Girschikofsky M, Arico M, Castillo D, et al. Management of adult patients with Langerhans cell histiocytosis: recommendations from an expert panel on behalf of Euro-Histio-Net. Orphanet J Rare Dis. 2013;14;8:72.
- Howarth DM, Gilchrist GS, Mullan BP, et al. Langerhans cell histiocytosis: diagnosis, natural history, management, and outcome. Cancer. 1999;85(10):2278–2290.
- Yi-Chin S, Wan-Lung L, I-Hsin S. Langerhans cell histiocytosis in Taiwan: a retrospective case series in a medical center. Dermatologica Sinica. 2009;27(2):93–102.
- Morimoto A, Ishida Y, Suzuki N, et al. Nationwide survey of single-system single site Langerhans cell histiocytosis in Japan. Pediatr Blood Cancer. 2010;54:98–102.
- Ng SS, Koh MJ, Tay YK. Cutaneous Langerhans cell histiocytosis: study of Asian children shows good overall prognosis. Acta Paediatr. 2013;102(11):e514–e518.